Primer-Blastの使い方

今日はプライマーが簡単にデザイン出来るツール、Primer-Blastの使い方を紹介していきます。
プライマーブラストを使いたいけれど、操作が全くわからないという方は参考にしてください。
なお、今回はprimer-blastの具体例として、大腸菌のヒートショックプロテイン遺伝子から500bpほどをクローニングして、相同組み替えでノックアウト体を作るという実験を背景にして設定を行います(詳しくは”シングルクロスオーバー”で検索するか、教科書で勉強してください)
Primer-Blastの使い方
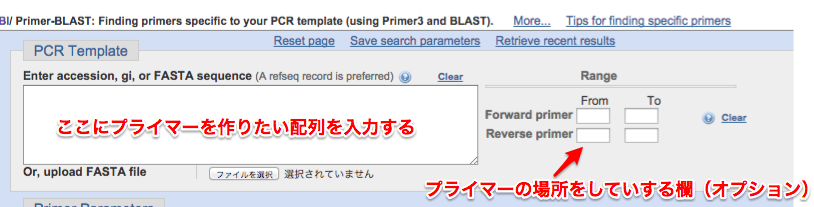
①http://www.ncbi.nlm.nih.gov/tools/primer-blast/にアクセス
②PCRテンプレートにプライマーで増やしたい領域を入力する。もしプライマーの場所で指定があるならば右のRangeで指定する。
具体例
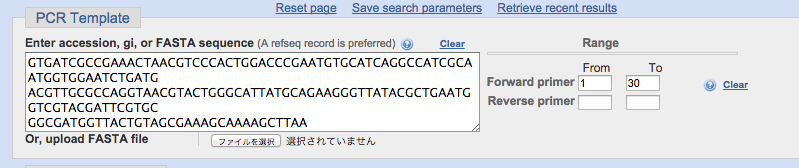
PCRテンプレートの欄に、大腸菌のヒートショックシャペロン(ibpB heat shock chaperone [ Escherichia coli str. K-12 substr. MG1655 ])の配列を入力しました。
また、ノックアウト用にクローニングするには遺伝子の上流がいいとされているので、前方30bp以内にフォワードのプライマーを設計するように指定しています。
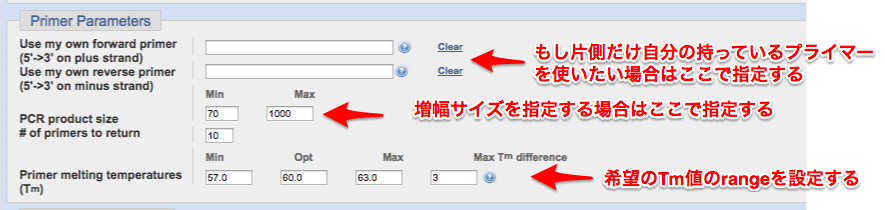
③ プライマーのパラメーターを設定していきます。
具体例
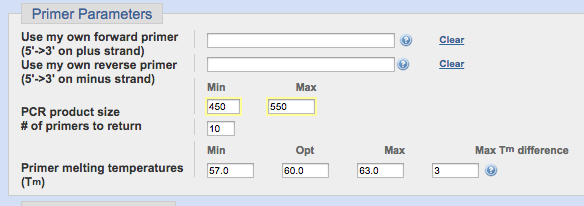
今回、増やしたい領域は500bp前後ですので、[PCR product size]は1割の幅を持たせて、450bpから550bpに設定しました。
Tm値は基本デフォルトの通り、60をOptとし、前後に3ほど幅を持たしておけば間違いはないです。
ただ、テンプレートの配列がGCリッチだったりとアブノーマルだと、この条件ではプライマーに最適な場所が見つからない場合がありますので、そのような場合は条件をもっと緩くしてみて下さい。
④ ExonJunction/Intronを噛ますかの設定をする
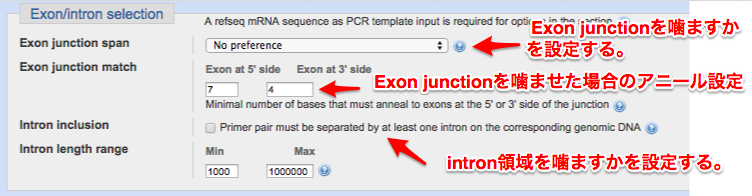
Tips
エキソンジャンクションとは、スプライシング後に出来るエキソンとエキソンの接合部を示します。
エキソンジャンクションを噛ませたプライマーを設計する事(下図参考)で、gDNAには存在しない配列のプライマーを設計することができ、gDNAからのPCR増幅を阻止することができます。
この方法は定量PCRの際に強力です。
何も考えずにプライマーを設計してPCRをすると、もしそのサンプルにgDNAがコンタミしていた場合には余計なものが増えてしまって正しい結果が得られません。
しかし、エキソンジャンクションを跨いだプライマーでしたら、gDNAにアニールすることはほとんどありませんので、gDNAがコンタミしたサンプルでも正しくmRNAを定量することが出来るのです。

具体例
今回のサンプルは大腸菌ですのでエキソン・イントロンはありません。
ですので、設定は全て”なし”で行います。
真核生物をお使いの方で、定量PCRをしたいと思っているかたはこのセクションを大事にしてください。
⑤ プライマーの特異性を確認する
※このセクションでは、設計したプライマーが対象物を特異的に増やすかを確認してくれます。例えば、遺伝子Aを増やしたいとしてプライマーを設計して、そのプライマーの配列が偶然にも遺伝子Bにも似ていたら、遺伝子A,B両方を増やしてしまう可能性があります。
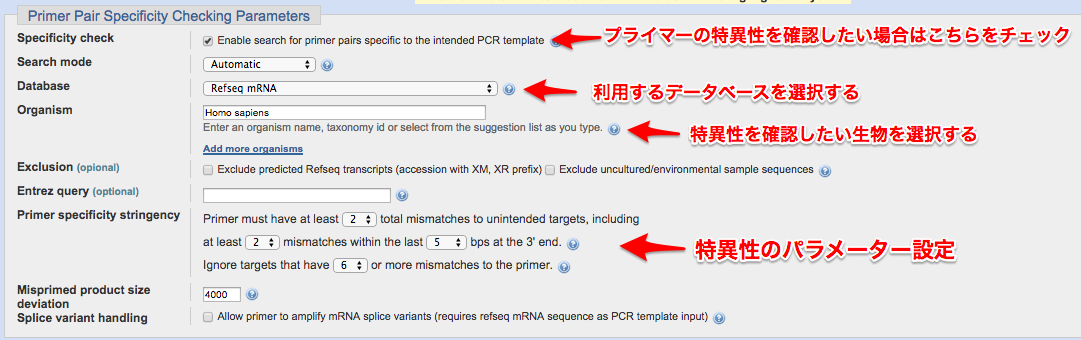
具体例
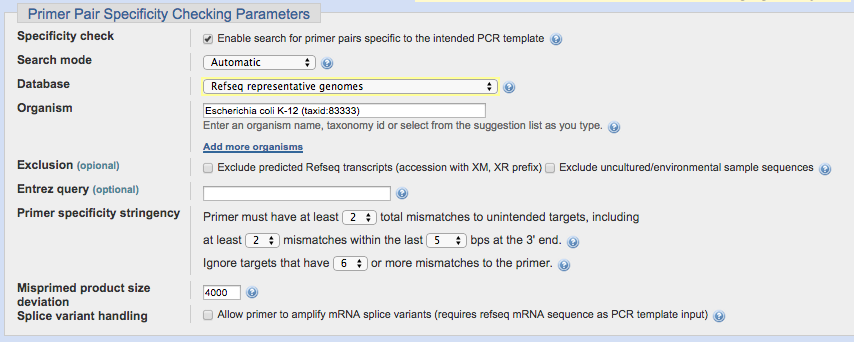
今回は、Escherichia coli str. K-12のゲノムデータベースを選択して、このヒートショックプロテイン用プライマーが同生物内の他遺伝子を増やさないかを確認します。
(Primer specificity stringencyのパラメーターはデフォルトで問題ありません)
結果
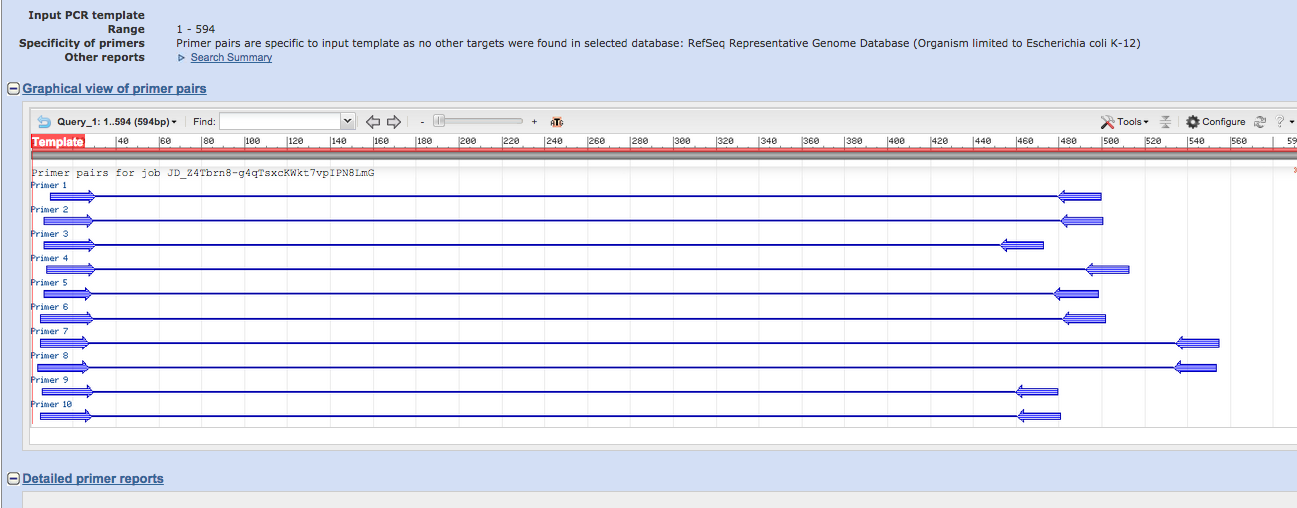
これがPrimer Blastが選んでくれたプライマーの場所になります。
この中から自分の好きなものを選んで使用しましょう。
では、これでPrimer Blastの使い方を終わります。

関連記事

コメント
Blast is not a good tool to check primer specificity. With these very short sequences a local alignment algorithm is not working very well. It s also a problem that you only can search public genomes. MFEprimer or other similar software works a lot better, and it can also be installed and run locally on propitiatory genomes.